
 Yahoo Autos
Yahoo Autos Human genome sequencing rises to the next level of diversity and accuracy

Twenty years after the first human genome sequence was published, an international research team has kicked the sequencing game to the next level with a set of 64 reference genomes that reflect much higher resolution and more genetic diversity.
Since the Human Genome Project completed the first draft of its reference genome, decoding the human genetic code has been transformed from a multibillion-dollar endeavor into a relatively inexpensive commercial service. However, commercial whole-genome sequencing, or WGS, often misses out on crucial variations that can make all the difference when it comes to an individual’s health.
“As a metric, 75% of structural variants that are present in that person’s genome are missed by WGS, but are captured by our long-read phased genome assembly,” University of Washington genome scientist Evan Eichler told GeekWire in an email. “Such variants are about three times more likely to cause disease.”
Eichler, who was a member of the original Human Genome Project, is one of the senior authors of a study laying out the new set of reference genomes, published today by the journal Science.
“Each of these individual genomes is being resolved more completely, for a fraction of the price of the first human genome,” he said in a news release. “We are discovering remarkable differences in genomic organization which have been missed until now.”
The Human Genome Project’s sequence — and another sequence that was published independently at the same time by a different group of researchers — were actually composites, produced by splicing together the genetic code from multiple individuals.
In contrast, the 64 newly published genomes document the precisely paired sets of maternal and paternal gene groups, or haplotypes, as reflected in genetic samples taken from 32 individuals. The individuals represent 25 different human populations from across the globe. Ten of the 32 samples came from people reporting African ancestry, who are typically underrepresented in genetic surveys.
“With these reference data, individual differences in terms of various types of genetic variants can now be studied with unprecedented accuracy,” said study lead author Peter Ebert, a researcher at the Institute of Medical Biometry and Bioinformatics at Heinrich Heine University in Germany.
Eichler told GeekWire that the improved understanding of the human genome “allows us to identify new hotspots of genetic instability that will be important for predicting where and why disease occurs — especially rare variants.”
“In addition to causing disease, structural variants are more likely to disrupt a gene function,” Eichler explained.
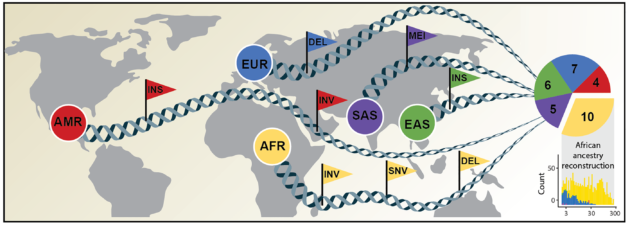
Researchers looked for examples where particular types of structural variants — for example, the insertion of hundreds of “letters” of genetic code — were more likely than other variants to affect gene expression. “This is not always a bad thing, and sometimes such changes are beneficial,” Eichler said. “Variants that are high in one human population versus another are good candidates.”
The sequencing effort resolved more than 100,000 structural variants, most of which were previously unknown.
Eichler said the analytical techniques used for the new reference genomes are likely to be a “game changer” for future genetic discoveries.
“It won’t happen tomorrow, but this is the way all human genomes will be sequenced clinically in the future,” he said in the news release. “Someday each person will have their individual human genome project to call their own, and having that information will improve their health.”
In addition to Ebert, the lead authors of the Science paper, titled “Haplotype-Resolved Diverse Human Genomes and Integrated Analysis of Structural Variation,” include Peter Audano of the University of Washington, Qihui Zhu of the Jackson Laboratory for Genomic Medicine and Bernardo-Rodriguez Martin of the European Molecular Biology Laboratory in Heidelberg. In addition to Eichler, the study’s senior and corresponding authors include Tobias Marschall of Heinrich Heine University, Jan Korbel of EMBL and Charles Lee of the Jackson Laboratory. In all, 65 researchers are listed as authors.
